Medizinprodukte. Software mit erhöhten Anforderungen.
Was versteht man unter Medizinprodukten? Welche Normen müssen Sie erfüllen und wie weist man deren Konformität nach?
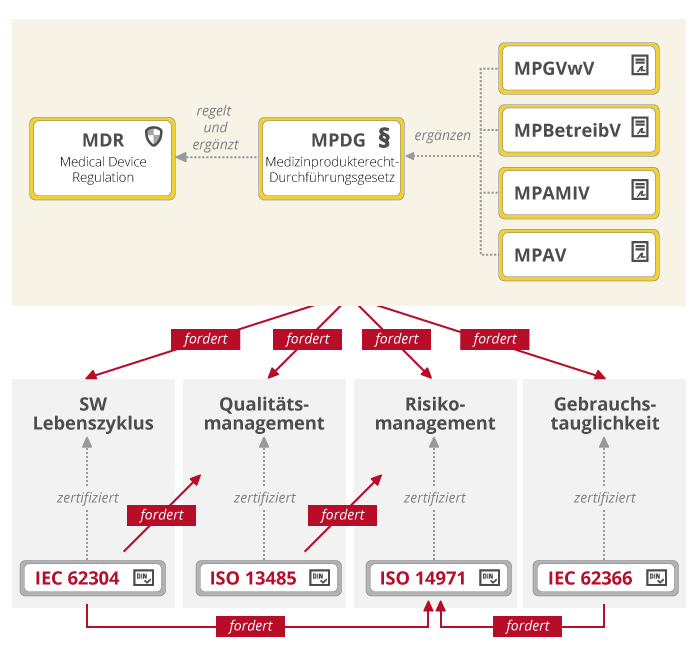
Die Medical Device Regulation (MDR) ist die Verordnung (EU) 2017/745 des Europäischen Parlaments und des Rates vom 5. April 2017 über Medizinprodukte.
Das deutsche Medizinproduktegesetz (MPG) wurde vom Medizinprodukterecht-Durchführungsgesetz (MPDG) abgelöst. Das MPDG gilt seit Mai 2021 schrittweise für alle Produkte im Anwendungsbereich der Verordnung. Es regelt und ergänzt die MDR.
Die harmonisierten Normen dienen den Herstellern zum Nachweis der Konformität mit den Richtlinien und nationalen Gesetzen.
Diverse Verordnungen gelten zusätzlich zur MDR in Deutschland.
Definition Medizinprodukte laut BGM
Laut Bundesministerium für Gesundheit sind Medizinprodukte Produkte mit medizinischer Zweckbestimmung, die vom Hersteller für die Anwendung beim Menschen bestimmt sind.
Dazu gehören Implantate, Produkte zur Injektion, Infusion, Transfusion und Dialyse, humanmedizinische Instrumente, Software, Katheter, Herzschrittmacher, Dentalprodukte, Verbandstoffe, Sehhilfen, Röntgengeräte, Kondome, ärztliche Instrumente, Labordiagnostika, Produkte zur Empfängnisregelung sowie In-vitro-Diagnostika.
Zu den In-vitro-Diagnostika zählen Reagenzien, Reagenzprodukte, Kits, Probenbehältnisse, Geräte und weitere Produkte, die zur In-vitro-Untersuchung von Proben aus dem menschlichen Körper bestimmt sind.
Medizinprodukte sind auch Produkte, die einen Stoff oder Zubereitungen aus Stoffen enthalten oder mit solchen beschichtet sind, die bei gesonderter Verwendung als Arzneimittel oder Bestandteil eines Arzneimittels (einschließlich Plasmaderivate) angesehen werden und in Ergänzung zu den Funktionen des Produktes eine Wirkung auf den menschlichen Körper entfalten können.
Anders als bei Arzneimitteln, die pharmakologisch, immunologisch oder metabolisch wirken, wird die bestimmungsgemäße Hauptwirkung bei Medizinprodukten primär auf physikalischem Weg erreicht.
Von der MDD zur MDR, vom MPG zum MPDG
Dem deutschen Medizinproduktegesetz übergeordnet war bis 2020 die europäische Medizinprodukterichtlinie Medical Device Directive MDD (93/42/EWG des Rates), die von der Verordnung über Medizinprodukte Medical Device Regulation MDR (EU) 2017/745 abgelöst wurde. Sie gilt ohne Umsetzung in nationale Gesetze seit dem 26.05.2021. Das Enddatum der Übergangsperiode für das erstmalige in Verkehr bringen von MDD Produkten ist der 26. Mai 2024. Die MDR wird als Verschärfung der MDD eingeschätzt, weil die Anforderungen an die klinische Bewertung wachsen. Die Klassifizierung der Produkte, die grundlegenden Anforderungen und die Konformitätsbewertungen blieben jedoch bestehen. Zusätzlich trat die Europäische Verordnung für In-vitro-Diagnostika IVDR mit der MDR am 25. Mai 2017 offiziell in Kraft. Die IVDR ist nach einer fünfjährigen Übergangszeit seit dem 26. Mai 2022 verpflichtend anzuwenden.
Das Medizinproduktegesetz (MPG) wurde 2020 vom Medizinprodukterecht-Durchführungsgesetz (MPDG) ersetzt. Das MPDG regelt und ergänzt die Anwendung der MDR (EU) 2017/745. Es ist durch Artikel 1 des Medizinprodukte-EU-Anpassungsgesetzes (MPEUAnpG) vom 28. April 2020 eingeführt worden.
Wann ist ein Medizinprodukt konform mit den Richtlinien? Wenn der Hersteller die Konformität mit den Richtlinien in einem Konformitätsbewertungsverfahren nachweist. Er erklärt das durch das Anbringen eines CE-Zeichens und benachrichtigt die im Gesetz benannten Stellen, dass er ein Medizinprodukt auf den Markt gebracht hat. Eine Zulassung oder Zertifizierung von Medizinprodukten gibt es in Europa nicht. Folglich kann man in Europa viel leichter Medizinprodukte in den Handel bringen als beispielsweise in den USA. Dort überwacht die FDA (Food and Drug Administration) die Zulassung von Lebensmitteln, Medikamenten und Medizinprodukten. Diese U.S. Behörde ist gefürchtet, weil sie nicht nur überwacht, sondern auch aktiv fahndet und mit aller Härte sanktioniert.
Umfrage von 2019 des Deutschen Industrie- und Handelskammertages (DIHK) und des Medizintechnik-Industrieverbands SPECTARIS.
Fast 80 Prozent der Medizintechnikunternehmen in Deutschland rechnen mit erheblichen Schwierigkeiten, innovative Produkte zukünftig auf den Markt zu bringen. Grund dafür sind MDR und IVDR.
Medizinprodukte-Verordnungen
Medizinprodukte-Durchführungsvorschrift
Die MPGVwV ist die allgemeine Verwaltungsvorschrift zur Durchführung des Medizinproduktegesetzes.
Medizinprodukte-Betreiberverordnung
In der MPBetreibV ist geregelt, wie der Betrieb von Medizinprodukten zu dokumentieren ist, wie und wie oft Kontrollen stattzufinden haben und welche Personen die Produkte bedienen und instand halten dürfen.
Medizinprodukte-Anwendermelde- und Informationsverordnung
In der Verordnung MPAMIV ist die Meldung von mutmaßlichen schwerwiegenden Vorkommnissen bei Medizinprodukten sowie zum Informationsaustausch der zuständigen Behörden geregelt.
Medizinprodukte-Abgabeverordnung
Die Verordnung MPAV regelt die Abgabe von Medizinprodukten.
Harmonisierte Normen
Die MDR und das MPDG definieren grundlegende Anforderungen an die Sicherheit von Medizinprodukten. Konkretisiert werden diese Anforderungen in puncto Qualität, Risiko, Usability etc. in sogenannten harmonisierten Normen:
ISO 13485: Medizinprodukte – Qualitätsmanagementsystem
ISO 14971: Anwendung des Risikomanagements auf Medizinprodukte
IEC 62304: Medizingeräte-Software: Software-Lebenszyklusprozesse
IEC 62366: Anwendung der Gebrauchstauglichkeit auf Medizinprodukte
IEC 60601-1: Medizinische elektrische Geräte
Die Anwendung dieser Normen ist nicht zwingend. Wenn sie jedoch nicht angewendet werden, muss auf andere Art nachgewiesen werden, dass das Medizinprodukt den Anforderungen entspricht. In der MDR ist zusätzlich von common specifications (CS) die Rede. Das bedeutet, dass die Hersteller in Zukunft weitere Beweisführungsinstrumente einsetzen müssen.
So erfüllen Sie Normen und Richtlinien in der Praxis
Erfahren Sie hier mehr über objectiF RPM – der Software für
Requirements Engineering und Projektmanagement »
Software as Medical Device (SaMD)
Bei Software im medizinischen Kontext unterscheidet man:
- Software als Teil eines Medizinprodukts z.B. als Embedded Software eines Medizingeräts,
- Software als eigenständiges Medizinprodukt (standalone Software),
- Software, die ein Zubehör zu einem Medizinprodukt ist und
- (eigenständige) Software, die kein Medizinprodukt ist.
Entscheidend ist die Zweckbestimmung des Herstellers. Weil diese Regelung aber ein wenig schwammig ist, hat die Medical Device Coordination Group (MDCG) eine neue Definition von Medical Device Software (MDSW) erarbeitet. Software als Medizinprodukt ist demnach sowohl Software, die unabhängig arbeitet als auch Software, die das Gerät zum Laufen bringt oder den Gebrauch beeinflusst.
Klassifizierung von Medizinprodukten
Medizinprodukte werden laut MDR in unterschiedliche Klassen eingeteilt unter Berücksichtigung der Zweckbestimmung der Produkte und ihrer inhärenten Risiken. Artikel 51 der MDR regelt die Zuteilung.
Klasse I: Geringes Risiko, sie werden in zwei Gruppen (steril und mit Meßfunktion unterschieden). Beispiele: medizinische Apps, Rollstühle, Brillen, Fieberthermometer etc.
Klasse IIa: Mittlerers Risiko. Beispiele: Zahnfüllungen, Röntgenfilme, Hörgeräte, Ultraschallgeräte etc.
Klasse IIb: Hohes Risiko. Beispiele: Intraokularlinsen, Kondome, Röntgengeräte, Infusionspumpen etc.
Klasse III: Sehr hohes Risiko. Beispiele: Hüft- und Kniegelenkimplantate, Herzkatheter, Brustimplantate etc.
Bei der Klassifizierung spielt eine Rolle, ob das Produkt invasiv ist, wie und wie lange die Anwendung stattfindet, ob es sich um ein aktives Produkt handelt und ob es an lebenswichtigen Organen eingesetzt wird. Sobald Software zu diagnostischen oder therapeutischen Zwecken eingesetzt wird (und das dürfte fast immer der Fall sein), wird sie je nach Risiko (Tod oder irreversible Veränderung des Gesundheitszustands) Klasse IIb oder sogar III zugeordnet.
Prof. Dr. Christian Johner:
Software ist ein Medizinprodukt, wenn der Hersteller sie zur Diagnose, Therapie oder Überwachung von Krankheiten und Verletzungen vorgesehen hat. Punkt.
IEC 62304: Software für Medizintechnik – Software-Lebenszyklusprozesse
Die Norm IEC 62304 benennt fünf Prozesse, die zum Lebenszyklus gehören:
- Software-Entwicklungsprozess
- Software-Wartungsprozess
- Software-Risikomanagementprozess
- Software-Konfigurationsmanagementprozess
- Problemlösungsprozess für Software
Der Entwicklungsprozess von Software wird in acht aufeinanderfolgende Schritte gegliedert:

Welche dieser acht Prozessschritte im Software-Entwicklungsprozess durchlaufen und dokumentiert werden müssen, hängt von der Sicherheitsklasse ab. Laut IEC 62304 gibt es drei Klassen Software:
Klasse A Verletzungen oder Gesundheitsschädigungen können durch den Einsatz der Software nicht auftreten.
Klasse B Schwere Verletzungen können durch den Einsatz der Software nicht auftreten.
Klasse C Verletzungen bis hin zum Tod sind durch den Einsatz der Software möglich.
Seit 2015 gibt es einen Zusatz zur Norm, in dem Software-System-Tests für alle Klassen vorgeschrieben sind. Was das Qualitäts- und Risikomanagement in der Softwareentwicklung anbelangt, verweist diese Norm auf die jeweils darauf spezialisierten Normen ISO 13485 und ISO 14971. Für die Wartung der Software gibt es einen ähnlichen Plan mit acht Schritten.

ISO 13485 Medizinprodukte – Qualitätsmanagementsysteme
Die Norm für die Qualitätssicherung von Medizinprodukten ISO 13485 ist in vielen Teilen mit der allgemein bekannten Qualitätsmanagement-Norm ISO 9001 identisch. Zentrale Forderung ist die Dokumentation aller Prozesse in einem Qualitätsmanagementhandbuch. Dieses Dokument bietet auch die Grundlage für den Konformitätsnachweis. In der ISO 13485 werden vier Prozessgruppen definiert:
- Die Verantwortung des Managements, das die Qualitätsziele definiert und das Qualitätsmanagementsystem auf seine Wirksamkeit überwacht.
- Das Management der Ressourcen (Personen, finanzielle Mittel und Ausstattung).
- Die Produktrealisierung einschließlich Entwicklung und Produktion.
- Der Prozess der kontinuierlichen Analyse und Verbesserung.
Diese Prozesse sollen dazu führen, die Anforderungen für regulatorische Zwecke vollständig zu erfüllen. Die Norm zielt in erster Linie auf die Sicherheit der Medizinprodukte ab, während die ISO 9001 darauf ausgerichtet ist, dass Organisationen eine kontinuierliche Verbesserung anstreben.
SO 14971 Anwendung des Risikomanagements auf Medizinprodukte
Die DIN ISO 14971 beschreibt einen Risikomanagementprozess für Medizinprodukte. Ziel dessen ist es, die Risiken für Patienten, Anwender und Dritte zu minimieren und ein akzeptables Nutzen-/Risiko-Verhältnis herzustellen. Die dritte Auflage der Norm, die im Dezember 2019 veröffentlicht wurde, betont noch stärker, dass der Nutzen eines Medizinproduktes die Risiken überwiegen muss. Dafür wurde Nutzen noch einmal genau definiert als:
positive Auswirkung oder wünschenswertes Ergebnis der Verwendung eines Medizinproduktes auf die Gesundheit einer Person oder positive Auswirkung auf das Patientenmanagement oder die öffentliche Gesundheit.
Mit Nutzen ist also ganz klar der medizinische Nutzen gemeint, nicht der wirtschaftliche Nutzen für den Hersteller.
Der Risikomanagementprozess beinhaltet sowohl die Risikoanalyse als auch das Risikomanagement. Auch dieser Prozess wird in mehrere Schritte unterteilt:
- Festlegen genereller Anforderungen an das Risikomanagementsystem
- Risikoanalyse
- Risikobewertung
- Risikobeherrschung
- Bewertung des gesamten Restrisikos
- Überprüfung des Risikomanagements
- Kontinuierliche Risikoanalyse und Verfikation der Risikoakzeptanz in der Produktion und Post-Produktion