Medical Products. Software with Increased Requirements.
What are medical products according to German law? What norms must they comply with and how is compliance determined?
Die Medical Device Regulation (MDR) ist die Verordnung (EU) 2017/745 des Europäischen Parlaments und des Rates vom 5. April 2017 über Medizinprodukte.
The Medical Product Law [Medizinproduktegesetz] transposed the MDD or MDR into German law. It was replaced by the Medical Devices Implementation Act (MPDG) in 2020. The MPDG has been progressively applicable to all products within the scope of the regulation since May 2021. For in vitro diagnostic devices, the Medical Devices Act was still applicable on a transitional basis until May 25, 2022.
The harmonized standards are used by manufacturers to demonstrate conformity with directives and national laws.
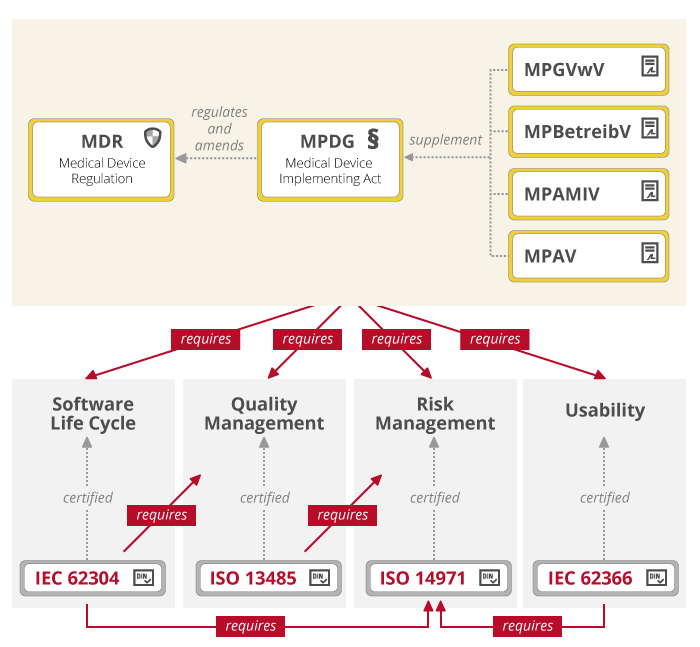
Various regulations apply in addition to the MDR in Germany.
As Defined by the Federal Ministry of Health (Germany, BGM)
Medical devices are products with a medical purpose that are intended by the manufacturer for use in humans. (Federal Ministry of Health)
These include implants, products for injection, infusion, transfusion and dialysis, human medical instruments, software, catheters, pacemakers, dental products, dressing materials, visual aids, X-ray equipment, condoms, medical instruments, laboratory diagnostics, products for regulating conception and in vitro diagnostics.
In vitro diagnostics include reagents, reagent products, kits, specimen containers, devices and other products intended for in vitro examination of specimens from the human body.
Medical devices also include products containing or coated with a substance or preparations of substances which, when used separately, are considered to be medicinal products or components of a medicinal product (including plasma derivatives) and are capable of exerting an effect on the human body in addition to the functions of the product.
Unlike medicinal products, which have a pharmacological, immunological or metabolic effect, the main intended effect of medical devices is achieved primarily by physical means.
From MDD to MDR, from MPG to MPDG
Until 2020, the European Medical Device Directive MDD (93/42/EEC of the Council) was superordinate to the German Medical Devices Act, which was replaced by the Medical Device Regulation MDR (EU) 2017/745. It applies without transposition into national laws since May 26, 2021. The end date of the transition period for the first placing on the market of MDD products is May 26, 2024. The MDR is assessed as a tightening of the MDD because the requirements for clinical evaluation are growing. However, the classification of products, essential requirements and conformity assessments remained. Additionally, the European In Vitro Diagnostics Regulation IVDR officially entered into force with the MDR on May 25, 2017. The IVDR has been mandatory since May 26, 2022, after a five-year transition period.
The Medical Devices Act (MPG) was replaced by the Medical Devices Law Implementation Act (MPDG) in 2020. The MPDG regulates and supplements the application of MDR (EU) 2017/745, and was introduced by Article 1 of the Medical Devices EU Adaptation Act (MPEUAnpG) of April 28, 2020.
When is a medical device compliant with the regulations? When the manufacturer proves conformity with regulations through a conformity assessment procedure. The manufacturer must affix a CE mark to the product and notify the bodies designated by law that a medical device has been placed on the market. There is no approval or certification of medical devices in Europe. Consequently, it is much easier to bring medical devices to market in Europe than in the USA, for example. There the FDA (Food and Drug Administration) oversees the approval of food, drugs and medical devices. This U.S. authority actively investigates and undertakes rigorous enforcement.
2019 survey by the Association of German Chambers of Industry and Commerce (DIHK) and Medical Technology Industry Association SPECTARIS.
Almost 80 percent of medical technology companies in Germany expect considerable difficulties in bringing innovative products to market in the future. The reasons for this are MDR and IVDR.
Harmonized Standards
The MDR and the MPDG define basic requirements for the safety of medical products. These requirements regarding quality, risk, usability etc. are specified in so-called harmonized standards:
ISO 14971: Application of risk management to medical products
IEC 62304: Medical device software: Software life cycle processes
IEC 62366: Application of usability to medical products
IEC 60601-1: Electrical medical devices
The application of these standards is not mandatory. However, if they are not applied, it must be proven by other means that the medical product meets the requirements. In MDR, common specifications (CS) are also mentioned. This means that manufacturers will have to use further instruments of proof in the future.
How to Meet Standards and Regulations in Practice
Learn more about objectiF RPM – the software for
requirements engineering und project management »
Software as Medical Device (SaMD)
There are several distinctions made for software in medical contexts:
- software as part of a medical product, e.g. as embedded software in a medical device
- software as a standalone medical product
- Software as an accessory to a medical product
- independent software that is not a medical product
The purpose as determined by the manufacturer is decisive here. However, because this regulation is a bit vague, the Medical Device Coordination Group (MDCG) has developed a new definition of Medical Device Software (MDSW). Software as a medical device is according to this definition both software that works independently and software that makes the device work or influences its use.
Medical Product Classification
Medical products are divided into different classes taking into account the intended purpose of the devices and their inherent risks.
Article 51 of the MDR governs the allocation.
Class I: Low risk, these are divided into two groups (sterile and with measuring function). Examples: medical apps, wheelchairs, glasses, thermometers, etc.
Class IIa: Medium risk. Examples: Dental fillings, X-ray films, hearing aids, ultrasound equipment etc.
Class IIb: High risk. Examples: Intraocular lenses, condoms, X-ray equipment, infusion pumps, etc.
Class III: Very high risk. Examples: Hip and knee joint replacements, heart catheters, breast implants etc.
Classification is based on whether the product is invasive, how and for how long it is used, whether it is an active product and whether it is used on vital organs. When software is used for diagnostic or therapeutic purposes (and this is likely always to be the case), it is assigned to Class IIb or even III, depending on the risk (death or irreversible change in health status).
Prof. Dr. Christian Johner:
Software is a medical device if the manufacturer intends it to be used for the diagnosis, treatment or monitoring of diseases and injuries. Period.
IEC 62304: Software for Medical Technology – Software Life Cycle Processes
Norm IEC 62304 names five software life cycle processes:
- software development process
- software maintenance process
- software risk management process
- software configuration management process
- problem solving process for software
The software development process is divided into eight sequential steps:

Class A Injuries or damage to health cannot occur through the use of the software.
Class B Serious injuries cannot occur by using the software.
Class C Injuries including death are possible as a result of using the software.
Since 2015 there is an addendum to the standard, in which software system tests are mandatory for all classes. As far as quality and risk management in software development is concerned, this standard refers to the respective specialized standards ISO 13485 and ISO 14971. For software maintenance there is a similar plan with eight steps.

ISO 13485 Medical Products – Quality Management Systems
The standard for quality assurance of medical devices ISO 13485 is in many parts identical with the widely known quality management standard ISO 9001. The central requirement is the documentation of all processes in a quality management manual. This document also provides the basis for proof of conformity. Four process groups are defined in ISO 13485:
- Management responsibility for defining quality objectives and monitoring the effectiveness of the quality management system.
- Resource management (people, financial resources and equipment).
- Product realization including development and production.
- The process of continuous analysis and improvement.
These processes were created to ensure full compliance with the requirements for regulatory purposes. ISO 13485 is primarily aimed at the safety of medical devices, while ISO 9001 is designed to ensure that organizations strive for continuous improvement.
ISO 14971 Application of Risk Managemant to Medical Products
DIN ISO 14971 describes a risk management process for medical products. The aim is to minimize the risks for patients, users and third parties and to establish an acceptable risk-benefit ratio. The third edition of the standard, which will be published in 2019, emphasizes even more strongly that the benefits of a medical device must outweigh the risks. To this end, benefit was precisely redefined as:
positive effect or desirable outcome of the use of a medical device on the health of a person or positive impact on patient management or public health
Benefit is thus clearly defined as the medical benefit, not the economic benefit for the manufacturer.
The risk management process includes both risk analysis and risk management. This process is also divided into several steps:
- Determination of general requirements for the risk management system
- Risk analysis
- Risk Assessment
- Risk management
- Evaluation of the total residual risk
- Review of risk management
- Continuous risk analysis and verification of risk acceptance in production and post-production